
Intestinal permeability is increased in patients with Crohn’s disease as well as some of their healthy first degree relatives. It has therefore been suggested that increased tight junction permeability (reduced barrier function) is a risk factor for development of Crohn’s disease. It is, however, not known if the healthy relatives with barrier loss, i.e. increased permeability, are at increased risk of developing disease. We tested this idea by generating a mouse in which intestinal paracellular, i.e. tight junction, permeability was increased due to intestinal epithelial-specific expression of a constitutively-active myosin light chain kinase (Su et al. 2009. Gastroenterology). Although these mice did display increased intestinal leak pathway permeability, they did not develop spontaneous disease. However, when challenged using an immune-mediated model of inflammatory bowel disease the mice developed more aggressive disease more quickly than mice without constitutively-active myosin light chain kinase and, ultimately, had reduced survival.
As discussed on a separate page, myosin light chain kinase primarily regulates the paracellular leak pathway. However, we have found that mice expressing constitutively-active myosin light chain kinase also have increased pore pathway permeability as a result of increased intestinal epithelial claudin-2 expression (Weber et al. 2010. J Biol Chem). Conversely, mice lacking the intestinal epithelial isoform of myosin light chain kinase are protected from immune-mediated colitis (Su et al. 2013. Gastroenterology). Eventually, these mice do succumb to disease as a result of apoptosis-driven, tight junction- and myosin light chain kinase-independent barrier loss. These data suggest that myosin light chain kinase inhibition may be therapeutic in maintaining remission or treating mildly active disease, but is unlikely to be beneficial in advanced, ulcerating disease. Myosin light chain kinase inhibition may however result in serious toxicities, and, as discussed on a separate page, other approaches will be needed.
Using intravital imaging of mice expressing EGFP-occludin within intestinal epithelia, we found that TNF-induced myosin light chain kinase activation causes tight junction barrier loss by activating caveolin-1-dependent endocytosis of occludin (Marchiando et al. 2010. J Cell Biol). While the function of occludin is controversial, we and others have shown that occludin knockdown in cultured monolayers results in increased leak pathway flux. We have gone on to show that TNF fails to induce occludin endocytosis in cells lacking ZO-1 or, alternatively, cells expressing occludin mutants with impaired ZO-1 binding (Buschmann et al. 2013. Mol Biol Cell). Ongoing studies will further define the molecular interactions that regulate occludin internalization and, more critically, why occludin removal from the tight junction increases leak pathway permeability.
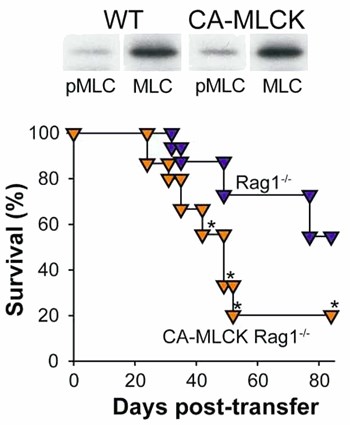
Transgenic constitutively-active myosin light chain kinase (CA-MLCK) expression within intestinal epithelial cells causes intestinal epithelial MLC phosphorylation, intestinal barrier dysfunction, and reduce survival in immune-mediated colitis. Phosphorylated MLC of WT and CA-MLCK Tg mice. P<0.01 (top). Survival after adoptive transfer of CD4+CD45Rbhi effector T cells into RAG1 knockout (blue symbols) and CA-MLCK transgenic RAG1 knockout recipients. Weight loss was far more severe (not shown) and survival was dramatically reduced by CA-MLCK expression. P<0.01 (bottom). From Su et al. 2009. Gastroenterology.
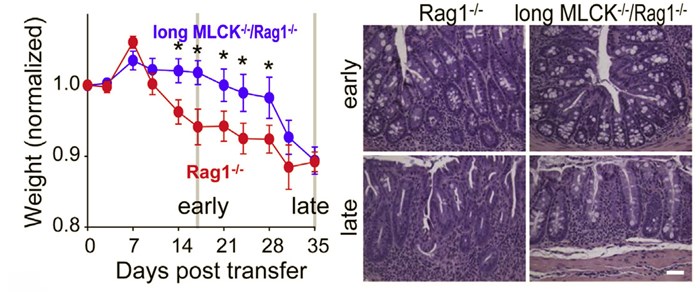
Long MLCK-/- mice are protected from CD4+CD45RBhi adoptive transfer colitis induced. Disease initiation and progression are delayed in long MLCK-/- mice in terms of weight loss (left) and colonic histopathology (right). Over time, weight loss in long MLCK-/- mice develops and is eventually comparable to that in MLCK-sufficient mice. This shift is associated with epithelial damage taht results in mysoin light chain kinase- and tight junction-independent barrier loss. In contrast, histopathology is less severe in long MLCK-/- mice even at late times. Bar = 0.1 mm. From Su et al. 2013. Gastroenterology.
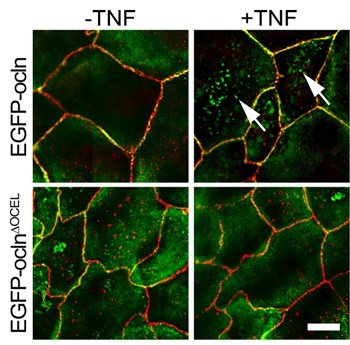
The occludin OCEL domain mediates binding to ZO-1 and is required for TNF-induced occludin endocytosis. TNF treatment induced EGFP-occludin, but not EGFP-occludinΔOCEL, internalization. EGFP-occludin–containing vesicles (green) were readily detected after TNF treatment (arrows). EGFP-occludinΔOCEL–containing vesicles (green) were not seen. ZO-1 (red) was detected by immunostaining. Bar, 10 μm. From Buschmann et al. 2013. Mol Biol Cell.
Jerrold R. Turner, MD, PhD
Professor of Pathology and Medicine
Brigham and
Women's
Hospital
Harvard
Medical
School
HNRB 752
77 Avenue Louis Pasteur
Boston, MA 02115
© Copyright JR Turner 2020.